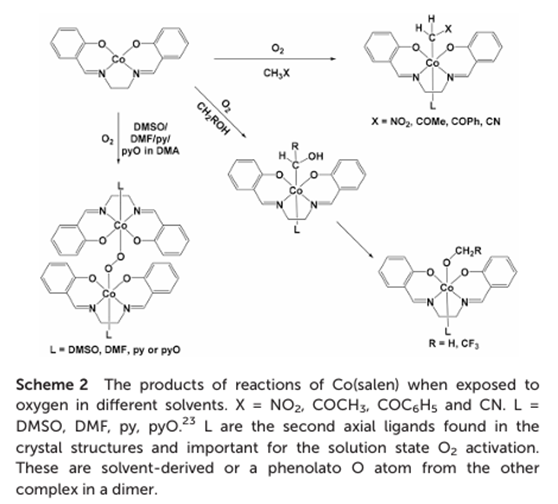
Dalton Trans.,2021,50,4819–4829一文中报道了二水杨醛缩乙二胺(Salen)钴(II)配合物在氧气存在下可以从多种溶剂(如丙酮、乙腈、硝基甲烷、醇类等)中攫取氢,并生成C–Co键,产率达到50% - 80%。反应条件为先回流15 min,此时溶液即由SalenCo的红色变成产物的深绿色;随后冷却并在黑暗中结晶2 d。除了硝基甲烷外,所有的底物都需要加入显著量的醇。
根据up主群青丨Ultramarine的实验视频,虽然回流过程在有光条件下进行,但在结束回流时,溶液底部仍然存在大量红色的SalenCo沉淀,它们在黑暗中会逐渐转变为绿色的产物晶体。因此,这个反应不太可能是光照导致的。
原文中对此反应机理进行了多种中间体表征和计算研究。其中,形如(MeOH)SalenCo(OO)的过氧配合物可以通过EPR鉴定,表现为一个二重态物种。
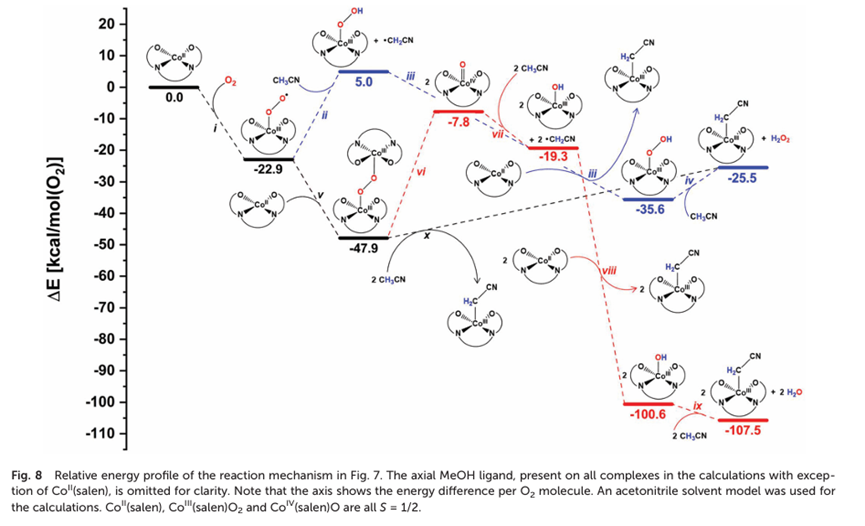
原文虽然对反应机理进行了计算,但呈现的结果十分令人困惑。根据这一结果,首先SalenCo与氧气会发生强放热的结合,这一反应的自由能变高达-22.9 kcal/mol,远远超过常理范围,且与已知CoSalen可逆地吸收和释放氧气的事实不符。此这一过氧自由基还会结合另一分子SalenCo,生成双核的SalenCoOOSalen物种,它将作为resting state,导致过氧自由基的HAT反应表观能垒高达52.9 kcal/mol。虽然文章 尬讨论了很多(例如作者认为超氧物种会先接触溶剂分子而不是发生二聚,但这毫无道理,因为一个能垒(注意全文中都将热力学与能垒混为一谈,此处使用‘能垒’一词是由于热力学规定了能垒的下限)至少为+27.9 kcal/mol的反应不可能同扩散竞争),但显然,根据所呈现的计算结果,双核物种将成为唯一可观测的产物,而后续HAT过程不可能发生;同样也不可能观测到(MeOH)SalenCo(OO)的EPR信号。这显然与实际情况不符。
为了澄清这些困惑,我们重新沿着最标准、规范的计算流程,重新探讨这个反应的机理。
1. 计算方法
虽然相关物种可能涉及较强的静态相关,但使用多参考态方法对这一反应进行全面的能量计算并不现实,而对于此类过渡金属配合物恰当选择的泛函则往往可以以相当低的花销得到令人满意的精度。首先进行快速的泛函测试,使用PBE0-D3BJ/def2-SV(P)/SMD(MeOH)水平下优化的SalenCo和(MeOH)SalenCo(O2)二重态结构,以及(MeOH)SalenCo(O2)四重态结构,以在NEVPT2(11,13)水平下得到二重态–四重态能量差作为测试指标。使用ORCA 6.1测试了60个泛函,均结合def2-TZVP基组,除了双杂化外,能用色散校正的都带D3BJ.
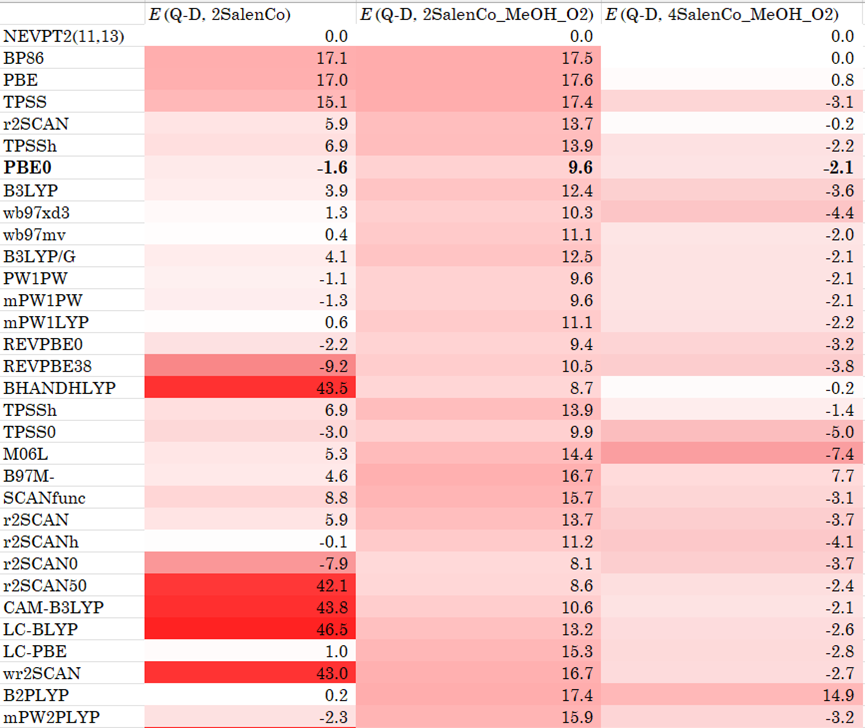
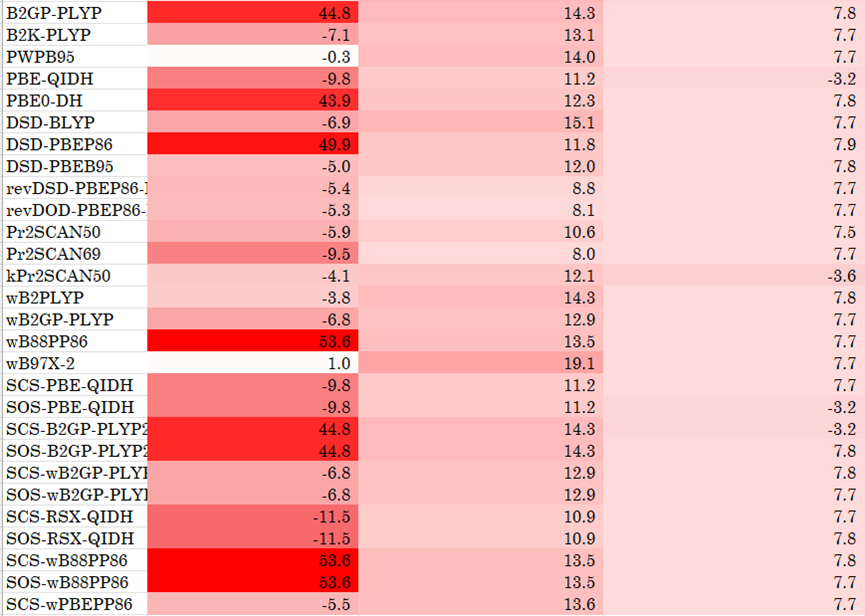
表1. 各泛函的测评数据,展示带符号误差(kcal/mol)。部分误差很大的可能由于波函数不稳定,但出于高通量筛选的目的,在此阶段没有对波函数稳定性进行进一步核对。
容易看出,PBE0在这个自旋态能量差问题上的表现脱颖而出,与一般认为PBE0适合过渡金属体系的经验也相符。wB97家族虽然也表现不错,但对SalenCo的四重态容易带有波函数不稳定性,出于便利性考虑不再采用。因此对后续即计算采用PBE0-D3BJ/def2-TZVP/SMD(MeOH)//PBE0-D3BJ/def2-SV(P)/SMD(MeOH)水平。构型优化、频率计算、单点计算使用Gaussian 16,298 K和1 mol/L下的自由能校正借助Goodvibes程序使用quasi-RRHO方法进行。甲醇的自由能通过气相甲醇及室温下甲醇的蒸汽压计算(这很重要)。
需要特别说明的是,这一部分提到的四重态–二重态能量差E(Q–D)都是针对二重态势能面上的极小几何结构而言的,不代表这个化合物采取何种基态。这也在后面的讨论中可以看出。
2. SalenCo的基本性质
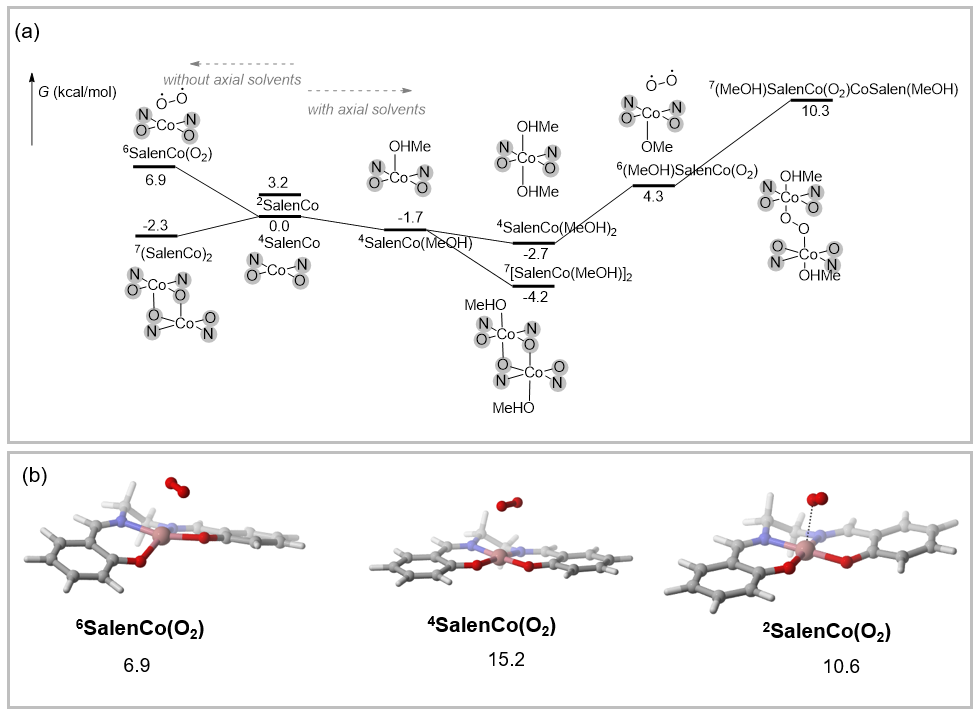
图1. (a) SalenCo在甲醇溶液中的配位和二聚行为自由能面,所有物种均展示自旋基态。 (b) SalenCo – O2复合物的结构和自旋态能量。
SalenCo相关的绝大多数物种在室温下的基态都是高自旋的。SalenCo自身的二聚是略微自发的(无甲醇时-2.3 kcal/mol,及有甲醇轴向配位时的-4.2 kcal/mol)。与原文中报道结果显著不同的是,SalenCo与氧气的结合导致自由能的显著上升,且产生的形式“超氧”配合物形成双核配合物更是明显吸能。这意味着溶液中SalenCo的主要存在形式是未结合氧气的SalenCo及其二聚体。
结合氧气后生成的SalenCo(OO)采取六重态基态。在六重态和四重态中,氧气与金属中心仅通过非共价作用结合,氧气维持三重态结构;仅二重态中存在一定的Co–O成键。轴向甲醇的存在并不影响该化合物的几何和电子结构特征。因此,虽然人们可以预期轴向强配体能通过稳定d6八面体场的方式稳定Co(III)OO·配合物,但显然甲醇的配位能力还远远不够。
与氧气的不利结合及不利的二聚解释了为什么不会从体系里分离出超氧配合物或双核配合物作为主要产物,也解释了为什么“超氧配合物”至多只能通过EPR表征。至于人们已知的当存在强配位性溶剂时可以沉淀出含有氧气的晶体,则依赖于晶体堆积效应在晶胞中恰好构筑了有利于氧气结合的刚性双钴位点。
既然甲醇不能通过轴向配位促进Co与氧气的结合,那它能不能通过氢键起到稳定相关中间体的作用呢?很遗憾,无论是与两个氧原子中的哪一个结合,相应物种的二重态(只在二重态下带有超氧或过氧特征)的生成都是进一步吸能的。
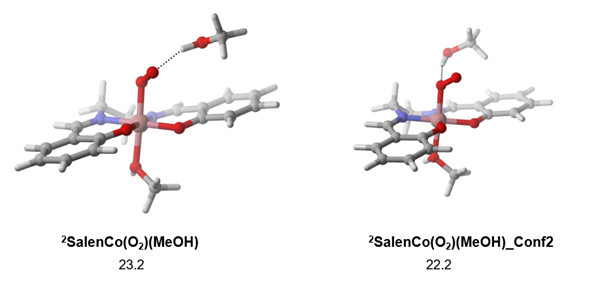
图2. 甲醇通过氢键结合时的SalenCo–O2复合物结构及相对自由能(kcal/mol)
综上,在这一部分,我们确认了以下结论:整个反应中不存在由于氧气结合或二聚带来的resting state;与氧气结合后的二聚吸能甚高,以至于仅从热力学就可以推定只有单核SalenCoOO配合物存在进行后续反应的可能。
3. “超氧”配合物与丙酮的HAT并无可能,及反应经超氧阴离子发生
在原文中,作者假设形如LSalenCoOO的“六配位超氧配合物”与丙酮、乙腈等发生HAT,以此作为Co – C键形成的起点。然而,简单的热力学考量就能否决这种观点:4(MeOH)SalenCo(O2)与丙酮生成1(MeOH)SalenCo(OOH)和相应碳自由基的自由能变高达30.2 kcal/mol!
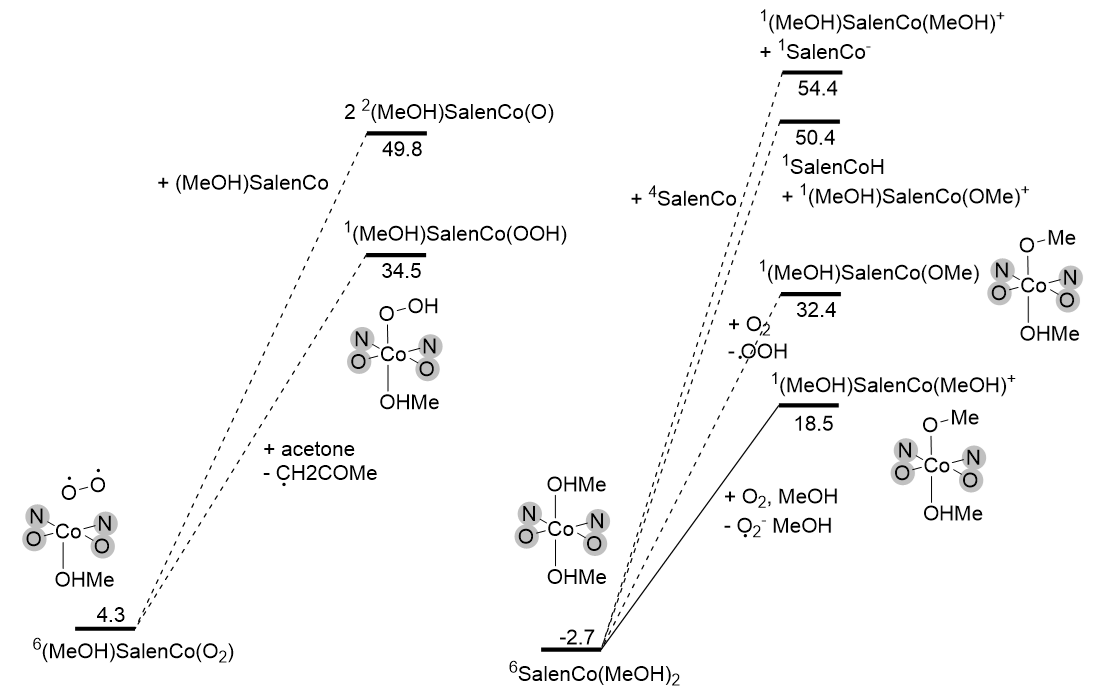
图3. 若干潜在反应途径自由能变(kcal/mol)
除了直接HAT外,其他潜在反应途径也被排除。“超氧”物种向另一分子SalenCo转移氧原子生成oxo-物种的自由能变超过45 kcal/mol;双甲醇配位的SalenCo被氧气直接攫取羟基氢生成过氧自由基和甲氧基Co(III)配合物、自身歧化生成Co(I)和Co(III)化合物,或是与甲醇发生双分子氧化加成得到Co(III)H和Co(III)OMe的热力学也都不可接受。而令人惊喜的是,SalenCo与氧气发生outer-shell电子转移,生成超氧自由基阴离子的过程在热力学上是允许的(以作为resting state的7[(MeOH)SalenCo]2为起点的相对自由能为22.7 kcal/mol)。考虑到这种电子转移通常不在热力学吸能的基础上带来多少附加能垒(虽然可以利用KST48程序方便地寻找交叉点来进行定量验证,但受时间所限我并不打算这样做) ,可以推测超氧自由基阴离子可以作为一种关键中间体。
一旦超氧自由基阴离子得以生成,通往产物的后续步骤可想而知都是快速且放热的,以至于对后续的详细计算也变得没有绝对的必要了。首先对丙酮进行去质子,生成的烯醇负离子随即与方才生成的Co(III)正离子结合得到产物;进一步与第二分子丙酮发生HAT,生成的碳自由基再与SalenCo结合生成第二分子产物(我们可能会关心OOH自由基是否会通过与SalenCo的结合而被淬灭,但不用担心,两者的结合仅放能1.9 kcal/mol)。生成的过氧化氢随即被SalenCo还原,生成羟基Co(III)物种和羟基自由基。羟基自由基可能会与丙酮反应,也可能再被SalenCo捕获。根据原文报道,这个羟基Co(III)物种可能是一种副产物或resting state,因为它在10 ℃时并不与乙腈反应,而在升温后则会重新转化为SalenCo(可能是通过Co – OH键的均裂)。如果羟基Co(III)物种对后续转化为惰性,那么以SalenCo计的总产率在50%到75%之间(取决于羟基自由基与丙酮反应和被捕获的相对速率);如果羟基Co(III)物种能缓慢地重新释放羟基自由基,则理论产率能达到100%。至此,我们对这个氧气活化-碳氢键活化反应的机理有了比较令人信服的认识。
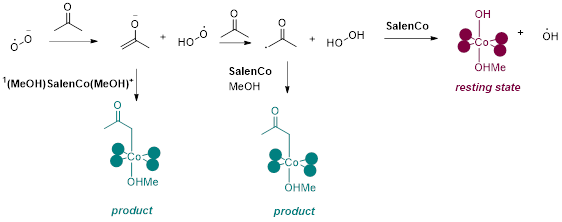
图4. 超氧阴离子向产物的转化