以下是去年出版的一本拙作的前两章内容。接下来我会逐渐更新计算化学的手把手系统教程,本章内容很适合作为先导,让读者知道计算化学究竟是干什么的,有什么意义。同时还顺便介绍了一些基本概念,以及从另一个角度推出自由能公式,也挺好玩的。
1.1 模型的建立和检验
在进入本书的讨论、借助计算化学的工具探索纷繁复杂的物质世界之前,首先提出一个问题:什么是科学研究?归根结底,科学家在做什么?
你可能会回答说,科学家进行日复一日的推理、演绎、实验、归纳,其目的是得到和增进对于客观世界的认识,从而实现对客观世界的掌握和操控。这个答案过于复杂了,我们能否用简单的几个字来概括科学研究的内容和目标?
回首自然科学的发展过程,我们很容易得到答案。现代化学脱胎于炼金术,在漫长的中世纪历史中,人们通过将不同的物质组合,在特定条件下进行反应,观察并记录了各种现象,但多数人都不认为炼金术是化学的一部分。通常认为,化学作为一门自然科学诞生于拉瓦锡和玻义耳的时代。玻义耳的《怀疑派的化学家》被认为是化学的开山之作,在其中他提出“化学,绝不是医学或药学的婢女,也不是甘当工艺和冶金的奴仆。化学本身作为自然科学中的一个独立部分,是探索宇宙奥秘的一个方面。化学,必须是为真理而追求真理的化学”。与此同时,他还发展了元素学说,并发展了理想气体定律。
在作为独立科学的化学诞生后,纵观其发展历史,从我们耳熟能详的重要事件中,我们很容易找到共性。1777年燃烧的氧气学说建立、1867年发现质量作用定律、1869年提出元素周期律、1874年提出碳的四面体结构、1916年提出路易斯结构式、1927年提出价键理论,所有这些决定了化学历史发展的标志性事件无一例外都与重要规律的发现、乃至于重要理论的建立相关。对于这些规律的认识深刻影响了人们看待物质世界的方式,每个化学家在此基础上进行思考,并通过自身的努力力求实现对规律的完善。
因此,我们可以发现,化学与炼金术不同、而作为一门科学的基础在于化学家的目的并非观察和记录现象,而是发现影响了物质变化行为的规律。而这些规律,都可以使用“模型”一词来描述。每种科学观点,都是对某些现象进行解释的一种模型。一言以蔽之,科学家的工作内容和目的,皆在于建立和检验模型。
模型可以通过多种方法得到。古人通过亲身体验和哲学思辨,认为冷热干湿四种属性决定了物质的行为,从而得到了四元素模型;近代的科学家通过对物质能否分解的观察,发现一些物质无法分解为其他物质并将其称为“元素单质”,从而建立了现代元素模型。Waage等人发现反应速率与浓度的幂成正比,总结为公式,形成了质量作用定律,构成了化学反应动力学的基本模型;后来人们发现许多反应的速率并不遵循质量作用定律,进一步提出这些反应由多种符合质量作用定律的基元反应构成。玻尔为了解释氢原子光谱,假定氢原子绕着原子核进行圆周运动,并提出了其原子结构模型;后来的科学家发现玻尔模型不能解释非氢原子的光谱,随后进一步提出了原子结构的量子力学模型。在这些例子中,我们能够发现科学研究的一些基本过程和方法,同时也是建立模型的基本过程和方法:观察、归纳、假设、演绎、检验。当一个模型能够解释当前观察到的所有现象时,它将被接受;当出现新的现象时,它可能依旧可以解释,从而得以保留,也可能无法解释,从而要么进行修正,要么被舍弃。
既然所有的科学研究过程都是在建立和检验模型,是不是只要提出一个能够解释该现象的模型就行?模型之间是否有高下之分呢?
让我们想象自己是一只猫。我们发现每过8小时,面前就会出现食物。作为猫中的科学家,通过观察归纳的方法,我们很容易建立这样的模型:在这个世界上,每隔8小时,食物就会自然出现。它完美地解释了当前观察到的所有现象,但从人类的视角来看,这个模型显然是荒谬的。如果这只猫坚持采用这个模型,当它的主人长期外出、并且忘记设置喂食时,这只猫就只能挨饿了。
仍然以猫举例。养过猫的人往往知道,晃动猫粮盒子时,猫就会跑过来。这是因为在猫看来,每次猫粮盒子发出声音后,就会紧跟着有食物倒出来。因此猫类科学家可能会得到这样的模型:猫粮盒子发出声音,会导致食物的出现。它同样解释了观察到的现象,但如果有一天主人只是为了让猫跑过来而晃动盒子、又不给它食物时,这只猫就会失望了。
上述两个模型都可以解释在猫的眼中观察到的事实,并且如果主人永远都循规蹈矩按时投喂,猫类科学家可能也永远观察不到与这些模型相冲突的现象。它可能会非常自豪,认为自己掌握了猫类科学的真理。但显然,这些模型都是错误的。
人类不应该嘲笑被困在家中的宠物猫。人类科学家发展出了纷繁复杂的手段,企图探究物质世界的规律,但这些努力在大自然面前仍然只是沧海一粟。人们既无法确保当前观察到的现象足够精密且不会出错,也无法预测未来会观察到怎样的现象,甚至有可能就像这只宠物猫一样受制于各种因素永远也无法观察到某些客观存在的现象。即使对于已经观察到的现象确定无疑,想要建立因果关系也十分困难,由于各种现象往往同时出现,难以将相关与因果分清,很难确保自己不会犯类似于“是盒子发出声音导致了食物出现”这种荒谬的错误。这些模型即使能够解释观察到的现象,也是由错误的原因偶然得到了正确的结论。对于科学研究来说,这甚至比无法得到确切结论更加有害,因为它会使得后人沿着错误的方向不断进发、投入越来越多的资源,当最终发现误入歧途时,往往已经走了相当多的弯路。
在科学发展的历史上,这种例子屡见不鲜。其原因多种多样。
第一种可能性是观察手段的欠缺。即使人们尽到了最大努力,由于各种检测手段时空分辨率的限制,仍然不能将现象背后的真正原因找到。就在最近(2021年),有人报道了一例无金属催化的Suzuki偶联反应)(Nat. Catal. 2021, 4, 71-78),并提出其采用的胺类催化剂独特的反应性导致了催化活性,一时引起了轰动。但很快,人们就发现,胺类催化剂并非起到决定性作用的物种,真正使得反应得以发生的是其中的痕量金属杂质。可以想象,如果人们没有发现真正的原因,想方设法去优化胺类催化剂的结构,将会面临多少不必要的困难。
第二种可能性是由于复杂现象可能有多种影响因素,彼此之间难以分离检验,很容易对现象产生错误的解释。例如,想象一个反应,与传统催化剂相比,某新型催化剂存在下表现出了良好的反应效果。有的人可能会很天真地提出解释:该催化剂有较高的催化活性,能够加速反应。但只需稍加思考,就能知道这种说法的不合理之处。针对观察到的现象,至少有如下几种解释:
与传统催化剂相比,新型催化剂能够加速主反应,并抑制副反应;
与传统催化剂相比,新型催化剂能够加速主反应,同时加速副反应,但对副反应的加速程度较少;
与传统催化剂相比,新型催化剂能够抑制主反应,同时抑制副反应,且对副反应的抑制程度较大;
与传统催化剂相比,新型催化剂对各反应速率没有什么影响,但有较好的稳定性;
与传统催化剂相比,新型催化剂对各反应速率没有什么影响,稳定性也差不多,但物料的传质更快;
与传统催化剂相比,新型催化剂对各反应速率没有什么影响,稳定性也较差,但溶解性更高;
……
可能对现象产生影响的因素彼此组合,有的产生积极影响,有的产生消极影响,在很多情况下,即使穷尽了充分的努力,也难以区分清楚究竟是哪一种情况。然而科学家的任务恰恰是找到其中的真相,如果对其原因进行了错误的指认,势必导致学术共同体在错误的道路上越走越远。
对于现象的错误归因带来的长期弯路的例子中,单分子磁体是一个典型。早期人们发现的单分子磁体往往是一些多核金属配合物,具有很高的自旋磁矩,因而人们很容易想当然地做出结论:自旋磁矩越高,单分子磁体的各项性能越好。然而经过20年的艰苦努力,不断试图寻找具有更高自旋磁矩的单分子磁体后,人们却发现鲜有突破,最终才恍然大悟,最初单分子磁体的性能与高磁矩的关系不过是巧合,甚至从某种意义上,高磁矩对于单分子磁体的稳定性是有害的。(Chinese Journal of Chemistry, 2020, 38(9): 1005-1018.)可见,当一个模型从错误的原因得到了“正确的”结果时,将产生很强的误导性,对科学发展有很大的危害。作为科学工作者,应当追求从正确的原因得到正确的结果;宁愿得不出结论,也应当设法避免对现象的错误归因。
经过上述的讨论,我们似乎陷入了一种困境。现象是无法穷尽的,观察是难以细致入微的,因此我们是否永远不可能知道这个世界的真正规律?
幸运的是,上述困境是建立在“我们通过观察和归纳认识世界”的基础上的。它之所以出现,正是由于观察永远是有限的,而且绝大多数实验观测都是通过所研究的目标物体对于外界的反应而进行的推论,因此都是间接证据;而在此基础上的归纳、对现象的解释更是在有限的间接证据基础上进行外推,难保不会出错。而除了观察和归纳外,我们还有其他的途径,即演绎和推理。
1.1 模型的建立和检验
在进入本书的讨论、借助计算化学的工具探索纷繁复杂的物质世界之前,首先提出一个问题:什么是科学研究?归根结底,科学家在做什么?
你可能会回答说,科学家进行日复一日的推理、演绎、实验、归纳,其目的是得到和增进对于客观世界的认识,从而实现对客观世界的掌握和操控。这个答案过于复杂了,我们能否用简单的几个字来概括科学研究的内容和目标?
回首自然科学的发展过程,我们很容易得到答案。现代化学脱胎于炼金术,在漫长的中世纪历史中,人们通过将不同的物质组合,在特定条件下进行反应,观察并记录了各种现象,但多数人都不认为炼金术是化学的一部分。通常认为,化学作为一门自然科学诞生于拉瓦锡和玻义耳的时代。玻义耳的《怀疑派的化学家》被认为是化学的开山之作,在其中他提出“化学,绝不是医学或药学的婢女,也不是甘当工艺和冶金的奴仆。化学本身作为自然科学中的一个独立部分,是探索宇宙奥秘的一个方面。化学,必须是为真理而追求真理的化学”。与此同时,他还发展了元素学说,并发展了理想气体定律。
在作为独立科学的化学诞生后,纵观其发展历史,从我们耳熟能详的重要事件中,我们很容易找到共性。1777年燃烧的氧气学说建立、1867年发现质量作用定律、1869年提出元素周期律、1874年提出碳的四面体结构、1916年提出路易斯结构式、1927年提出价键理论,所有这些决定了化学历史发展的标志性事件无一例外都与重要规律的发现、乃至于重要理论的建立相关。对于这些规律的认识深刻影响了人们看待物质世界的方式,每个化学家在此基础上进行思考,并通过自身的努力力求实现对规律的完善。
因此,我们可以发现,化学与炼金术不同、而作为一门科学的基础在于化学家的目的并非观察和记录现象,而是发现影响了物质变化行为的规律。而这些规律,都可以使用“模型”一词来描述。每种科学观点,都是对某些现象进行解释的一种模型。一言以蔽之,科学家的工作内容和目的,皆在于建立和检验模型。
模型可以通过多种方法得到。古人通过亲身体验和哲学思辨,认为冷热干湿四种属性决定了物质的行为,从而得到了四元素模型;近代的科学家通过对物质能否分解的观察,发现一些物质无法分解为其他物质并将其称为“元素单质”,从而建立了现代元素模型。Waage等人发现反应速率与浓度的幂成正比,总结为公式,形成了质量作用定律,构成了化学反应动力学的基本模型;后来人们发现许多反应的速率并不遵循质量作用定律,进一步提出这些反应由多种符合质量作用定律的基元反应构成。玻尔为了解释氢原子光谱,假定氢原子绕着原子核进行圆周运动,并提出了其原子结构模型;后来的科学家发现玻尔模型不能解释非氢原子的光谱,随后进一步提出了原子结构的量子力学模型。在这些例子中,我们能够发现科学研究的一些基本过程和方法,同时也是建立模型的基本过程和方法:观察、归纳、假设、演绎、检验。当一个模型能够解释当前观察到的所有现象时,它将被接受;当出现新的现象时,它可能依旧可以解释,从而得以保留,也可能无法解释,从而要么进行修正,要么被舍弃。
既然所有的科学研究过程都是在建立和检验模型,是不是只要提出一个能够解释该现象的模型就行?模型之间是否有高下之分呢?
让我们想象自己是一只猫。我们发现每过8小时,面前就会出现食物。作为猫中的科学家,通过观察归纳的方法,我们很容易建立这样的模型:在这个世界上,每隔8小时,食物就会自然出现。它完美地解释了当前观察到的所有现象,但从人类的视角来看,这个模型显然是荒谬的。如果这只猫坚持采用这个模型,当它的主人长期外出、并且忘记设置喂食时,这只猫就只能挨饿了。
仍然以猫举例。养过猫的人往往知道,晃动猫粮盒子时,猫就会跑过来。这是因为在猫看来,每次猫粮盒子发出声音后,就会紧跟着有食物倒出来。因此猫类科学家可能会得到这样的模型:猫粮盒子发出声音,会导致食物的出现。它同样解释了观察到的现象,但如果有一天主人只是为了让猫跑过来而晃动盒子、又不给它食物时,这只猫就会失望了。
上述两个模型都可以解释在猫的眼中观察到的事实,并且如果主人永远都循规蹈矩按时投喂,猫类科学家可能也永远观察不到与这些模型相冲突的现象。它可能会非常自豪,认为自己掌握了猫类科学的真理。但显然,这些模型都是错误的。
人类不应该嘲笑被困在家中的宠物猫。人类科学家发展出了纷繁复杂的手段,企图探究物质世界的规律,但这些努力在大自然面前仍然只是沧海一粟。人们既无法确保当前观察到的现象足够精密且不会出错,也无法预测未来会观察到怎样的现象,甚至有可能就像这只宠物猫一样受制于各种因素永远也无法观察到某些客观存在的现象。即使对于已经观察到的现象确定无疑,想要建立因果关系也十分困难,由于各种现象往往同时出现,难以将相关与因果分清,很难确保自己不会犯类似于“是盒子发出声音导致了食物出现”这种荒谬的错误。这些模型即使能够解释观察到的现象,也是由错误的原因偶然得到了正确的结论。对于科学研究来说,这甚至比无法得到确切结论更加有害,因为它会使得后人沿着错误的方向不断进发、投入越来越多的资源,当最终发现误入歧途时,往往已经走了相当多的弯路。
在科学发展的历史上,这种例子屡见不鲜。其原因多种多样。
第一种可能性是观察手段的欠缺。即使人们尽到了最大努力,由于各种检测手段时空分辨率的限制,仍然不能将现象背后的真正原因找到。就在最近(2021年),有人报道了一例无金属催化的Suzuki偶联反应)(Nat. Catal. 2021, 4, 71-78),并提出其采用的胺类催化剂独特的反应性导致了催化活性,一时引起了轰动。但很快,人们就发现,胺类催化剂并非起到决定性作用的物种,真正使得反应得以发生的是其中的痕量金属杂质。可以想象,如果人们没有发现真正的原因,想方设法去优化胺类催化剂的结构,将会面临多少不必要的困难。
第二种可能性是由于复杂现象可能有多种影响因素,彼此之间难以分离检验,很容易对现象产生错误的解释。例如,想象一个反应,与传统催化剂相比,某新型催化剂存在下表现出了良好的反应效果。有的人可能会很天真地提出解释:该催化剂有较高的催化活性,能够加速反应。但只需稍加思考,就能知道这种说法的不合理之处。针对观察到的现象,至少有如下几种解释:
与传统催化剂相比,新型催化剂能够加速主反应,并抑制副反应;
与传统催化剂相比,新型催化剂能够加速主反应,同时加速副反应,但对副反应的加速程度较少;
与传统催化剂相比,新型催化剂能够抑制主反应,同时抑制副反应,且对副反应的抑制程度较大;
与传统催化剂相比,新型催化剂对各反应速率没有什么影响,但有较好的稳定性;
与传统催化剂相比,新型催化剂对各反应速率没有什么影响,稳定性也差不多,但物料的传质更快;
与传统催化剂相比,新型催化剂对各反应速率没有什么影响,稳定性也较差,但溶解性更高;
……
可能对现象产生影响的因素彼此组合,有的产生积极影响,有的产生消极影响,在很多情况下,即使穷尽了充分的努力,也难以区分清楚究竟是哪一种情况。然而科学家的任务恰恰是找到其中的真相,如果对其原因进行了错误的指认,势必导致学术共同体在错误的道路上越走越远。
对于现象的错误归因带来的长期弯路的例子中,单分子磁体是一个典型。早期人们发现的单分子磁体往往是一些多核金属配合物,具有很高的自旋磁矩,因而人们很容易想当然地做出结论:自旋磁矩越高,单分子磁体的各项性能越好。然而经过20年的艰苦努力,不断试图寻找具有更高自旋磁矩的单分子磁体后,人们却发现鲜有突破,最终才恍然大悟,最初单分子磁体的性能与高磁矩的关系不过是巧合,甚至从某种意义上,高磁矩对于单分子磁体的稳定性是有害的。(Chinese Journal of Chemistry, 2020, 38(9): 1005-1018.)可见,当一个模型从错误的原因得到了“正确的”结果时,将产生很强的误导性,对科学发展有很大的危害。作为科学工作者,应当追求从正确的原因得到正确的结果;宁愿得不出结论,也应当设法避免对现象的错误归因。
经过上述的讨论,我们似乎陷入了一种困境。现象是无法穷尽的,观察是难以细致入微的,因此我们是否永远不可能知道这个世界的真正规律?
幸运的是,上述困境是建立在“我们通过观察和归纳认识世界”的基础上的。它之所以出现,正是由于观察永远是有限的,而且绝大多数实验观测都是通过所研究的目标物体对于外界的反应而进行的推论,因此都是间接证据;而在此基础上的归纳、对现象的解释更是在有限的间接证据基础上进行外推,难保不会出错。而除了观察和归纳外,我们还有其他的途径,即演绎和推理。
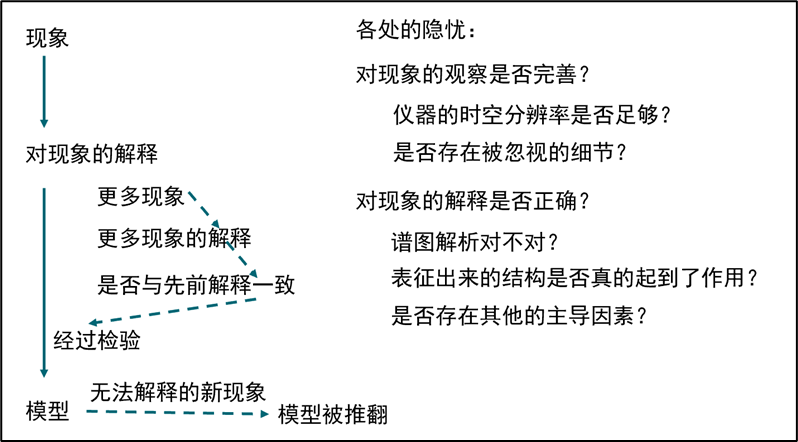
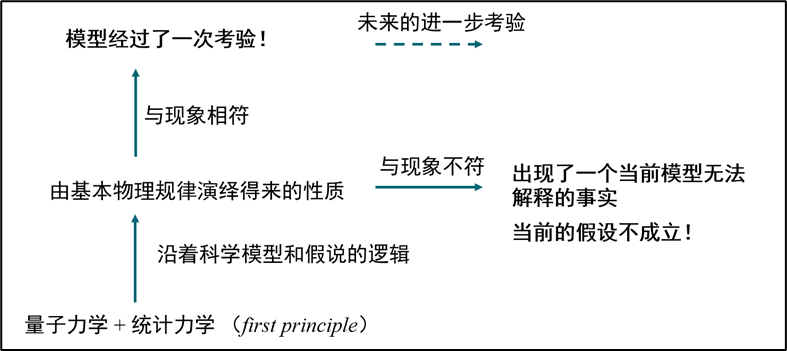
图1.1.1 基于观察和归纳的模型建立过程,以及基于演绎的模型建立过程
在这一过程中,我们首先承认一种广泛成立的基本原理。它在本质上同样是模型,但它具备当前人类认识水平中最高的抽象程度,经受过最广泛的检验,从而被认为在当前认识水平下处处成立的普遍规律。从这种基本原理出发,按照模型进行演绎,来看是否能够得到各种观察到的现象。如果两者一致,则该模型通过了这次检验,否则就意味着这种模型与最底层的普遍规律相冲突,需要对模型假设进行修正。在这种框架下,对模型检验的整个过程都不依赖于对现象的解释;正相反,它是针对“对现象的解释”本身来进行检验,恰恰非常适合检查模型的正确性。在化学世界中,这种广泛成立的基本原理是量子力学和统计力学。基于量子力学和统计力学的原理,从基本物理规律出发来构建对于化学世界的描述,进而检验各种因素对现象究竟有何种影响,是计算化学的任务。
使用这种从下向上的方法进行模型检验,有如下优点:
- 从原理上没有不确定度,因为所有结果都基于基本物理原理,仅受到基本物理常数(如光速、元电荷等)的影响。而在实际操作中采取的近似处理,所带来的误差范围对于讨论化学问题也十分足够。
- 与光谱等基于分子对外界响应进行推论而带来的间接证据不同,计算化学直接研究微观粒子的行为,相当于直接“看到”结果,属于直接证据。而受限于不确定性原理的限制,人们不可能从实验上以足够的精确度“看到”电子。
- 通过演绎推理,可以很好地将可能影响复杂现象的各种因素分离开,单独研究某种因素所起到的角色。想要验证哪个假设,就去计算相关的物理量,不受其他因素的干扰,指哪打哪。
以之前提到的催化反应为例,虽然多种可能的因素难以通过对实验现象的观察来区分,使用计算化学的方法就可以带来深入和全面的理解:
为了验证催化剂是促进还是抑制了主反应,只需对主反应的机理进行计算,得到能垒,以判断各种催化剂对反应的促进或抑制作用及其强度。
为了验证催化剂是促进还是抑制了副反应,只需罗列可能的副反应并依次计算其能垒。
为了验证催化剂的稳定性,只需罗列可能的催化剂分解途径并以此计算能垒。
为了验证催化剂的溶解度,可以尝试考察溶剂化能、晶格能等相关物理量。
在排除各种可能性之后,如果仍有未尽之处,可以推测存在传质传热等非化学因素起到了作用。
由此可见,计算化学的手段在检验模型方面有巨大的威力。通过本书的介绍,读者将能体会到这种威力,并初步理解如何利用计算化学排除错误的模型和假设、建立正确的科学观点,实现“从正确的原因得到正确的结果”。
1.2 物质结构:分子、晶体与表面
对结构的理解是化学家认识世界的基础,对结构的表征鉴定是化学家日常工作中最重要的任务之一。在计算化学中,对结构的正确认识更是至关重要。在计算化学的工作流程中,首先构建特定的结构,随后研究该结构的物质具备的性质,只有对结构有充分的把握,使得所研究的结构能够充分反映所研究的问题,才能得到有意义的结果。
各种物质随着状态和性质不同,其结构千差万别。化学家感兴趣的物质通常有如下几类:气相分子或离子;溶液;晶体、无定形固体及其表面。它们又可以大致分为两类:具有明确且有限的原子组成的分子(又称为孤立体系),和在微观尺度看来几乎是无限延伸的凝聚体(又称为周期性体系)。
各种固体是周期性体系的典型代表。对于晶体,我们将其视作由晶胞在若干方向上无限重复构成,这也正是“周期性体系”名称的由来。晶体的实际尺寸可以相当庞大,而只需研究其重复单元,就可以充分理解其各种性质。
考虑一块宏观晶体的边界,此处即为它的一处表面。虽然宏观晶体可以有多个表面,通常每处表面的大小都远超过了晶胞的尺寸,在微观尺度上,可以将表面视作由在二维方向上无限重复的晶胞构成,因此表面也是典型的周期性体系。
无定形固体与晶体不同,不存在严格意义上的晶胞,整体呈现出无序性。但为了研究方便,通常仍然可以视作由特定的晶胞重复构成,而在这一晶胞内人们可以通过各种方法将其原子排列打乱,构建能够反映其无定形特征的结构模型,因此通常也作为周期性结构处理。
与固体相反,气态的化合物由明确的分子或离子构成。除了在通常条件下呈现气态的物质外,在以质谱条件为代表的特定环境下,可以人为生成许多气相分子、离子或团簇。这些物质均属于孤立体系。例如虽然氧化铝在通常状态下是高熔点的固体,属于典型的周期性体系,但在气相下,则以由几个到几十个原子构成的分子团簇的形式存在(10.1039/B212654K),并且其形状与固体结构可能有很大的差异。
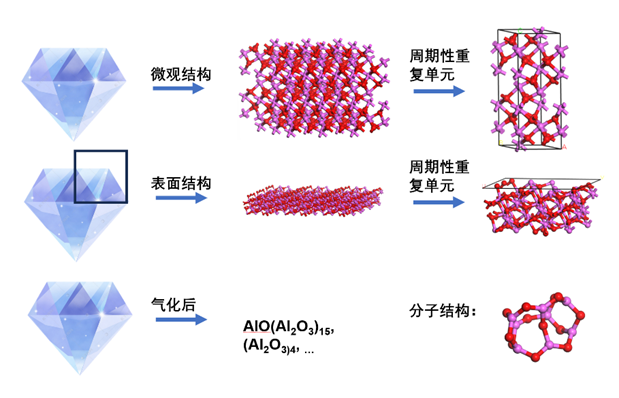
液体与固体类似,属于凝聚态,但无序程度相当高,组成溶液的各分子排列非常灵活。如果要研究溶液的实际结构,即其中各溶剂和溶质分子是如何排列的,通常仍然会使用周期性边界条件:建立适当大小、包含特定数量溶剂和溶质分子的盒子,并将体相溶液视作该盒子在三维方向无限重复构成。而如果并不关心溶液的实际结构,仅关心其中溶质的行为,则可以将溶液等效为一个外部环境,仅研究浸泡在这一外部环境中的溶质分子或离子,此时则成为了典型的孤立体系。
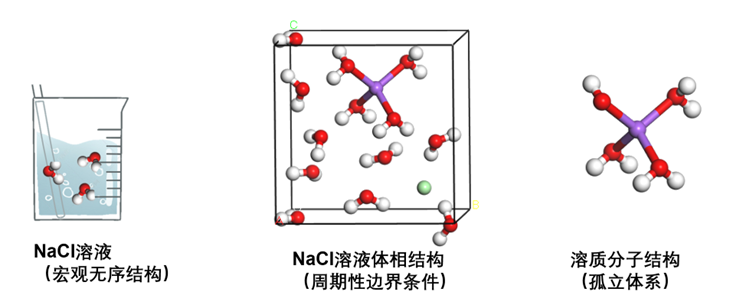
图1.2.2 溶液的体相和分子结构示意
大多数情况下,当研究溶液中的化学反应时,关注的都是溶质分子的变化,取溶质分子作为孤立体系进行研究是非常方便、准确的。在这一过程中,务必对溶液中分子的存在形态有充分的理解。以NaCl溶液为例,如要研究水中钠离子与15-冠-5(15-c-5)分子的结合情况,则需要知道NaCl在水中的存在形式为Cl-和Na(H2O)4+,且水中盐类完全电离,阴阳离子无相互作用,因此只需研究如下分子(离子)间的反应,即可得到该结合过程的自由能变:
Na(H2O)4+ + 15-c-5 → Na-15-c-5+ + 4H2O
在后续的章节中,我们将会知道,孤立体系与周期性体系适用于不同的处理方法,分别属于量子化学和第一性原理计算的范畴。
1.3 化学反应热力学
从广泛成立的基本原理出发构建化学世界的过程中,我们既要知道单个微观粒子的性质,这需要借助量子力学;也需要知道一群微观粒子的性质,这需要借助统计力学;还需要知道这些性质如何与宏观行为联系在一起,这需要借助化学反应热力学和动力学的知识。化学反应热力学和动力学是统计力学的推论,但由于其特殊的重要性,格外需要每个化学研究者关注并深入掌握。任何物理化学教科书中都会详细探讨化学反应热力学和动力学,在本节和接下来一节中,我们对其中要点进行回顾。
化学反应热力学探究了一个化学反应是否有可能发生、至多能以何种程度发生的问题,其中最重要的是平衡的概念。它解答了化学反应达到平衡时各物质的浓度是多少的问题。
化学反应热力学中最重要的概念是自由能。我们采用如下脉络,来重新梳理一遍自由能的定义。
在日常生活中,为了判断一个物体是否有向其他状态转变的趋势,我们从直觉上习惯通过受力来进行分析。当物体受力为0时,可以保持当前状态。在这其中又有稳定和不稳定两种情况。当一个正方体用一个面立在桌子上时,受合力为0,且保持稳定;而当它用一个顶点立在桌子上、且重心与顶点的连线垂直于地面时,其所受合力同样为0,但并不稳定,稍受扰动就会转变为用面立在桌子上的状态。
然而在化学世界中,我们讨论物质之间的转化时,所提到的物质几乎都可以自身稳定存在,这反映在受力上,就意味着各分子所受的合力始终为0。因此,无法将日常生活中基于受力分析建立起的朴素观念套用在化学世界中。取而代之,我们会通过能量来讨论。
能量需要满足如下性质:每个体系都能定义一个确定的能量;体系倾向于采取能量最低的状态;要想让体系的能量升高需要从外界得到能量。接下来让我们思考,如何定义一个满足这些性质的“能量”?
为了回答这个问题,自然而然地,我们会想到传热过程。几乎所有化学反应都伴随着热效应,传热过程的方向性容易与“能量降低”的方向联系起来。然而,不存在任何办法能够测定一个体系“包含多少热量”,只能测定出一个过程中两个相互接触的体系之间传递了多少热,因此热是与过程相关的量,不能满足“每个体系均能定义一个能量”的要求。但我们可以采取如下定义:
选定一个参考体系A,定义其属性X = 0.当它转变为体系B时,向外界传递了数量为Q的热,则体系B的属性X = -Q. 当这一过程伴随着做功时同理。通过这种方法,就定义了相对于某个参考基准的属性,满足我们期待“能量”能满足的性质。上述属性X,在恒容情况下被称作内能(U),在恒压情况下被称作焓(H)。
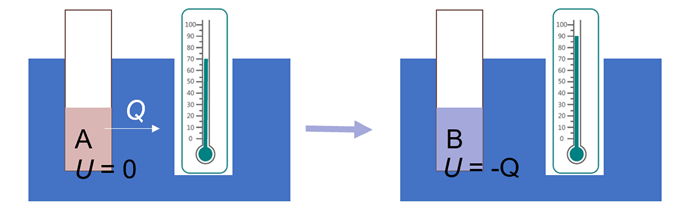
图1.3.1 通过热的传递衡量体系内能
接下来我们思考,上述定义的“能量”是否足以反映化学问题,也就是说,它是否使得我们足以相信,物质会倾向于采取使得这种“能量”最低的状态?
让我们回到日常生活中,很容易就会发现这种定义的不足之处。将体系的演化类比为一个在曲面上滚动的小球,显然小球倾向于停留在曲面上的洼地,从而使得重力势能取得最低。我们将重力势能类比为上述方式定义的“能量”,当小球停留在每个洼地所在的区域时,就认为它对应于某种状态。但接下来我们考虑这个问题:在地面上有两张地毯,都是平铺,故高度相同;两张地毯的面积不同,分别为A1和A2。 当小球落在某个地毯上时,就相当于进入了某种状态。当小球从高处随机滚落时,显然小球落在两个地毯上的概率不同,概率之比等于A1/A2。这个例子告诉我们,重力势能并非决定小球进入何种状态的唯一因素;在势能之外,还有一个统计因素在发挥作用。现在我们将势能项和统计项(暂时记作Y)相加,定义最终的能量E = H + Y或E = U + Y。
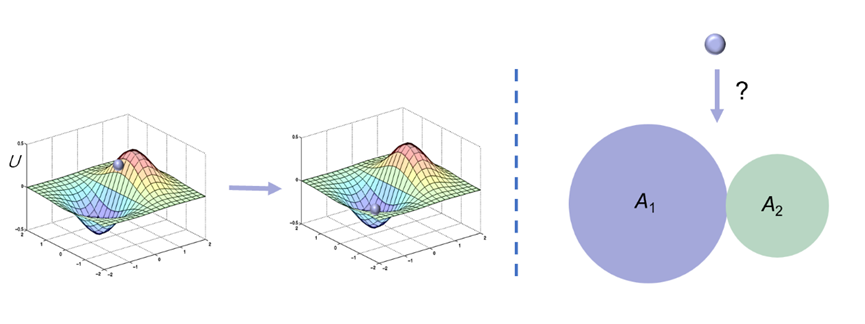
图1.3.2 化学反应的地毯小球模型
为了进一步确定这一统计因素的形式,引入玻尔兹曼分布,它可以通过经典物理学推导出来。即,对于一个正确定义的能量E,体系在平衡时处于状态1和状态2的概率比应当等于exp(-(E1 – E2)/kT),其中k为玻尔兹曼常数,T为温度。采用地毯上的小球模型,可得A1/A2 = exp(-(E1 – E2)/kT),又知H1 = H2,容易知道(Y1 – Y2) = -kTln(A1 – A2). 从而可以定义Y = -kTlnA。在化学世界中,地毯的面积对应了某个(宏观)状态内包含多少微观状态数N,这样我们就知道了决定体系平衡时状态的能量应当遵循H(或U) - RTlnN的形式,这一能量被称为自由能。定义熵S = klnN,可将自由能定义如下:
吉布斯自由能G = H – TS
亥姆霍兹自由能A = U – TS
它们完全满足我们对于能量的期待。自由能对于处于特定状态的体系唯一确定,并且仅与体系状态有关;体系倾向于降低其自由能,并且处于不同自由能的状态之间的概率比满足玻尔兹曼分布。这最后一点非常重要,因为它告诉了我们化学反应达到平衡时的浓度情况。由此可以得到关于平衡常数的结论:
对于每个化合物,可以定义其标准自由能。以化学反应aA + bB →cC + dD为例,标准自由能变为:
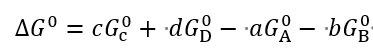
平衡常数由标准自由能变决定,为:
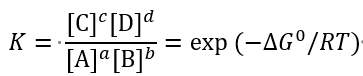
其中[C]等为各物质的平衡浓度,R为理想气体常数。因此,只要知道了一个反应的标准自由能变,就能知道达到平衡时各物质的含量,这是研究化学反应时最关键的信息之一。对于A→B的反应,当∆G‡ 为0时,表明平衡时A和B的含量相等,各占一半;如果∆G0 = +1 kcal/mol, 则室温下A的平衡浓度大约是B的5.4倍;如果∆G0 = -1 kcal/mol,则室温下B的平衡浓度大约是A的5.4倍。一种常见的说法是∆G‡ 表明了反应是否自发,显然,采取这一说法时必须严格限定“自发”的含义,绝不能理解为“∆G0>0的反应不能自行发生”。
自由能变是容量性质,与化学方程式的写法有关。通常能接触到的(即方程式中的计量数符合习惯、不太大或太小)大部分化学反应的自由能变在零点几到几十个kcal/mol的范围内。
自由能由焓和熵两项组成,焓的减少和熵的增加都有利于自由能的降低。其中焓与热效应有关,容易理解,而熵是统计效应,缺乏日常生活中的对应,因此我们通过地毯小球模型,尽可能希望帮助读者理解。对于分子,熵包含平动、转动和振动的贡献,分子越灵活、运动越自由,能采取的状态就越多,具备越高的熵。其中平动熵对于大多数分子数量级相似,大约为30 e.u.,其中1 e.u. = 1 cal/(mol·K)。在2分子结合为1分子的过程,1各分子的平动熵被部分损失了,并且结合越强,损失的熵越多。因此,几乎所有结合过程都伴随着明显的熵减。当焓效应不足以抵消熵减时,就会发现结合自由能为正值。例如众所周知水分子非常倾向于形成氢键,然而用热导率法测定出水分子形成氢键二聚体的焓变和熵变分别为-3.63 kcal/mol和-18.61 e.u. (10.1016/0009-2614(78)85290-7),容易知道这一结合过程在373 K下的自由能变为+3.3 kcal/mol。这在基于非共价作用的结合中是普遍现象。
1.4 化学反应动力学
化学反应热力学回答了反应体系在平衡时各物质的浓度问题,但没有回答需要如何才能达成平衡的问题。发生反应并达到平衡需要时间,化学反应的速率随着反应本性不同而跨度很大,某些高度活泼的中间体能在几百飞秒内发生反应,而许多反应等待几百万年也难以发生。化学反应动力学解答了与化学反应的速率有关的问题。
构成化学反应过程的基础是基元反应。基元反应是那些通过分子的一次或几次(由于不一定每次碰撞都有足够恰好的能量来激活反应)碰撞、或化学键的一次或几次(与碰撞同理)振动就可以发生的反应,除此之外的被称为复杂反应,是一系列基元反应的组合。反应机理即为构成复杂反应的基元反应的序列。以乙酸乙酯水解为例,总反应为MeCO2Et + H2O → MeCO2H + EtOH,机理可能如下:
MeCO2Et + H3O+ → MeC(OH+)OEt + H2O
MeC(OH+)OEt + H2O → MeC(OH)(OH2^+)(OEt)
MeC(OH)(OH2+)(OEt) + H2O → MeC(OH)(OH+)(OEt) + H3O+
MeC(OH)(OH+)(OEt) + H3O+ → MeC(OH)(OH)(OHEt+)
MeC(OH)(OH)(OHEt+) → MeC(OH+)OH + EtOH
MeC(OH+)OH + H2O → MeCO2Et + H2O
(以上展示的是特殊酸催化的机理,还可能有一般酸催化、特殊碱催化、一般碱催化、proton-shuttle等其他机理)。
分子反应中的基元反应通常有如下几类:
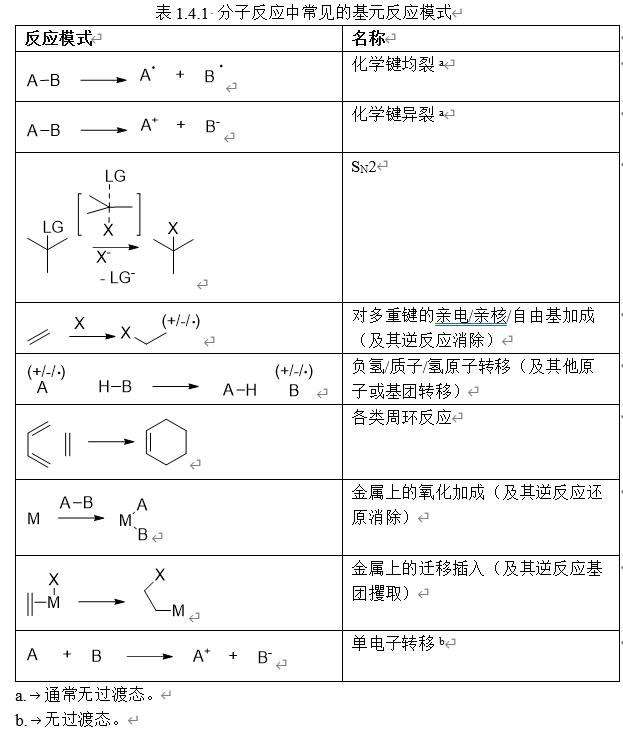
表面反应的机理往往更为复杂,表面大量悬挂键的存在使得其电子结构变化多端,而且这些反应经常在较高温度下进行,使得许多非常规的反应模式得以发生,基元反应的种类比较丰富。
定义反应速率r为浓度随时间的变化率。形如aA + bB →cC + dD的基元反应的速率遵从质量作用定律:
r=k[A]a [B]b
可见基元反应的速率正比于反应物浓度的幂,指数恰好等于发生该基元反应中该分子的计量数,称作反应对该物质的级数。显然,基元反应对某物质的级数要么是0,要么是正整数。k是与反应本性和温度有关的常数,称作速率常数。
只有基元反应严格服从质量作用定律。对于复杂反应,其表观速率可通过其机理、结合各基元反应的质量作用定律推导而来。它可能仍然服从质量作用定律的形式,也可能是许多服从质量作用定律形式的项之和,也可能更为复杂。对于服从质量作用定律形式的反应,可以类似地定义其对各物质的级数,这些级数既可能是整数也可能是分数,既可能是正数也可能是零或负数。
例如丙烯与氯化氢的加成反应,其表观速率方程表现出两项之和的形式:
r= k_1 [alkene] [HCl]2+ k_2 [alkene][HCl]
这提示该反应有两种机理共存,其中一条对氯化氢为一级,另一条对氯化氢为二级。
基元反应的速率常数服从Eyring方程:
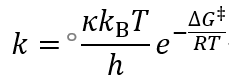
其中h为普朗克常数,kB为玻尔兹曼因子,T为温度。κ为传输因子,对每个基元反应为一个确定的常数。在其中包含一个玻尔兹曼形式的指数项,∆G‡ 称作活化自由能(Gibbs free energy of activation),又称活化能垒(Gibbs free energy barrier)。与化学反应热力学中的自由能类似,活化自由能也可以分解为活化焓和活化熵,它们统称为活化参数。
活化自由能的物理意义与过渡态的概念相关。在势能面上,每个化合物都是能量的极小值点,对其结构的微小扰动都将导致能量上升。在一个基元反应过程中,随着反应进行,绝大多数情况下能量并非在底物和产物能量之间单调变化。在反应的初期,随着底物结构发生扭曲重组,能量逐渐上升;随后底物逐渐向反应物转变,产生的新的相互作用使得能量下降,在反应路径上产生一个能量的极大值点。沿着反应路径方向的能量极大值点被称为过渡态(transition state, TS)。为了抵达过渡态而需要付出的能量构成了活化自由能。
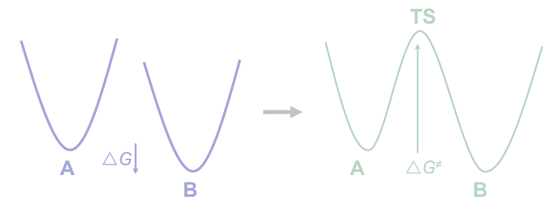
图1.4.1 反应路径上的过渡态与活化自由能
由于过渡态不是极小值点,分子不会在过渡态停留可观测的时间,没有任何方法能够直接观测过渡态的结构。想要探讨过渡态的结构和性质,可以通过一些机理研究手段进行间接推测(见下一节),而如果想要直接观察过渡态的几何和电子结构,必须借助计算的手段。
对于复杂反应,表观速率常数与温度通常仍然表现出与Eyring方程类似的指数关系,此时可以定义表观能垒(overall barrier)。通过Eyring方程,我们可以估算室温下表观能垒与一级反应速率常数和半衰期的关系。
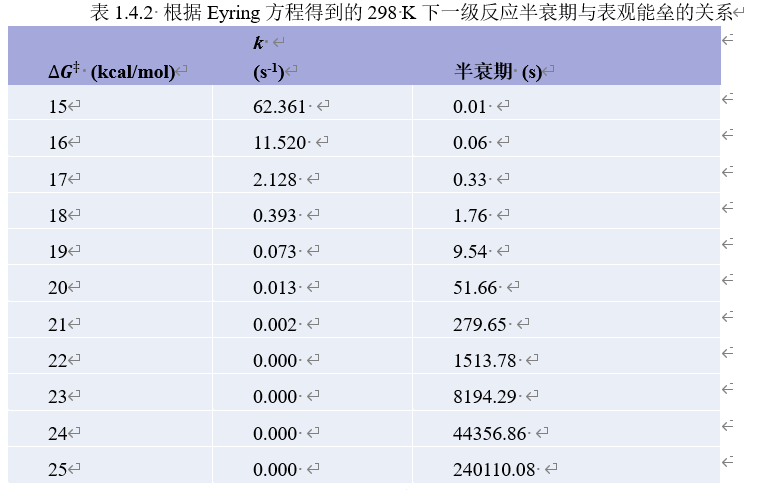
可见反应速率随能垒改变而显著变化,室温下能垒每升高1 kcal/mol,速率就降低大约5倍。表观能垒低于23 kcal/mol的一级反应可以认为能够在室温下在可接受的时间尺度(几小时到几天)内发生。通常,在讨论计算结果时我们可以将这条界限放宽到25 kcal/mol。用类似的方法,可以估计其他温度下能垒在何种范围内的反应能以可观的速率发生。
1.5 机理和研究机理的手段
对反应机理的探究和解析是化学研究的重要主题,也是对化学反应进行深入认识和理性设计的基础。机理研究主要有以下手段:
- 反应级数和活化参数
动力学是机理的直接体现,对动力学的测定和分析是研究机理最重要的手段。其中最重要的是反应级数和活化参数。反应级数可以通过准一级条件下测定特定物质浓度随时间的关系确定,也可以通过测定初始速率随初始浓度的关系确定。活化参数可以通过变温条件下的反应速率测定。通过反应级数的研究,可以判断哪些分子参与到了决速步和决速步之前的步骤中,以及它们的当量;而活化参数则可以帮助判断决速步过渡态的性质,特别是活化熵可以用于判断决速步中分子数量的变化。如以1.4节中所举的烯烃与氯化氢反应为例,其动力学方程包含对氯化氢为一级和二级的两项,提示该反应可能存在两种机理共存,且在决速步中分别有一分子和两分子氯化氢参与。这对应了烯烃亲电加成的两种机理:
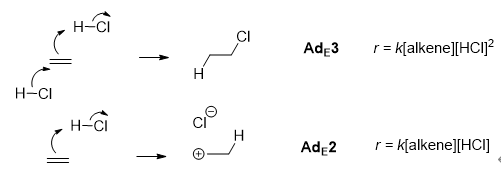
图1.4.2 烯烃亲电加成的两种典型机理
- 同位素实验
动力学同位素效应(kinetic isotopic effect, KIE)的测量构成了同位素实验中最重要的部分。KIE实验中,对某个原子进行同位素取代,研究同位素带来的速率变化,以判断该原子在决速步中的角色。最常见的是氢原子KIE,当决速步中涉及某个氢原子与其他原子之间的化学键断裂时,用氘取代氢,将导致明显的速率减慢(正常的一级KIE);类似地,当决速步中不直接涉及这个氢原子的变化,但氢原子所连接的原子的杂化方式改变时,将产生较小的二级KIE。
除了氢原子KIE外,随着核磁共振技术的进步,对碳原子或其他重原子KIE的测量技术也得到了发展,并能够带来多种机理信息。
除了KIE,同位素取代也能带来更多独特的机理信息。由于同位素的化学性质大体上相同,通过在反应物的特定位置引入同位素,观察产物中的同位素分布,可以追踪相关原子在反应过程中的去向。在一些反应中,原子位置可能随着一些可逆过程的不断发生而改变,此时通过观察同位素的分布是否发生紊乱,可以判断机理中的某些步骤是否可逆。
- 中间体捕获
当怀疑某些中间体参与到了反应过程中时,可以尝试通过分离纯化、加入捕获剂、或采用原位光谱手段,对该中间体的存在性进行验证。如果能将该中间体分离出来,再投入到反应体系中,发现该中间体可以顺利转化为产物,则可以在一定程度上支持它在反应过程中的角色。
需要注意的是,能够分离捕获的中间体往往有较高的稳定性,并非所有中间体都能分离。此外,捕获到该中间体只能证明其存在于反应体系中,而不能证明它在反应过程中究竟起到了何种角色。即使该中间体能够在反应条件下转化为产物,也无法证明它是直接出现在反应路径上,还是需要先转化为其他中间体再发生反应。因此,中间体捕获只能是证明“相关性”的手段,而对“因果性”的证明,必须结合更多更充分的证据。
- 机理计算
与之前各种手段不同,计算化学的手段不是通过反应体系表现出的特征进行推测来间接研究机理,而是直接得到拟定出来的机理中各步的热力学和动力学,从而验证该机理是否可行。其具体过程将在第7章进行探讨。机理计算是用于检验模型的手段,在进行机理计算之前,必须尽可能收集各种证据,拟定出若干有可能的机理,这就需要综合运用上述提到的各种机理研究的方法,并充分考验着化学家的智慧和劳动。而在此基础上,计算化学带来的直接与定量检验机理模型的能力,则极大地增强了化学家对化学反应机理的把握和信心。