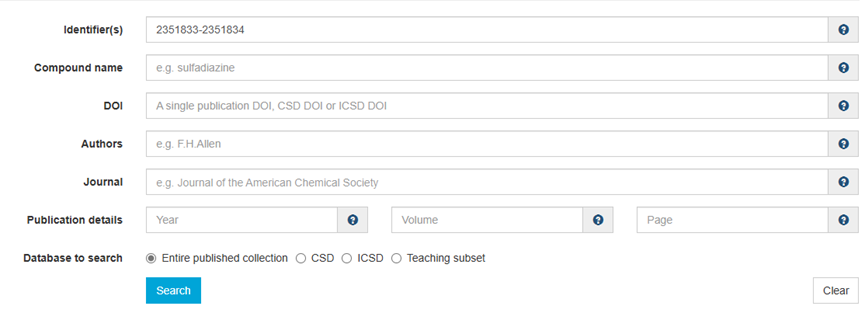
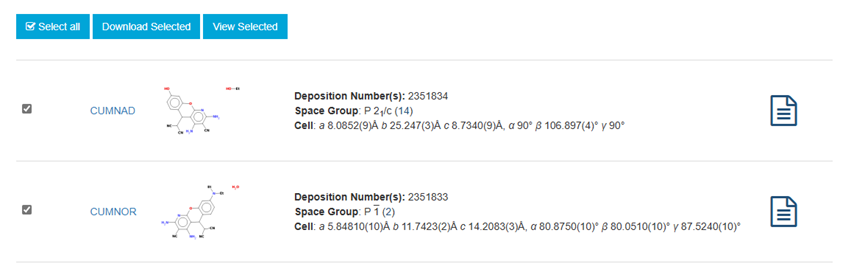
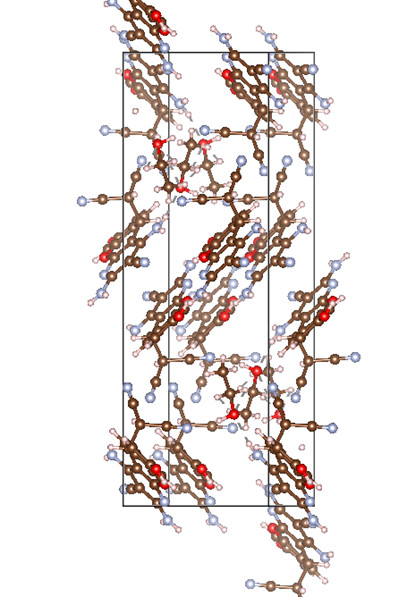
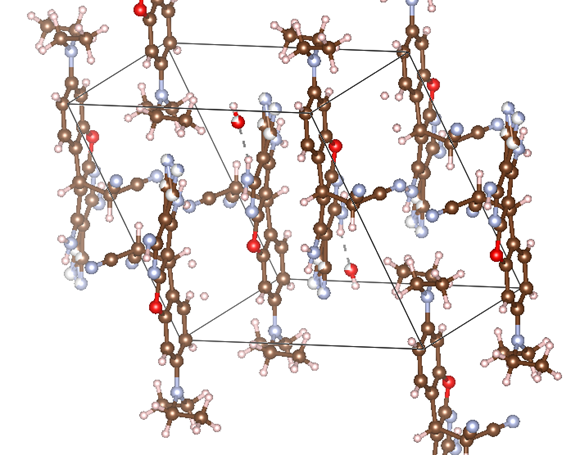
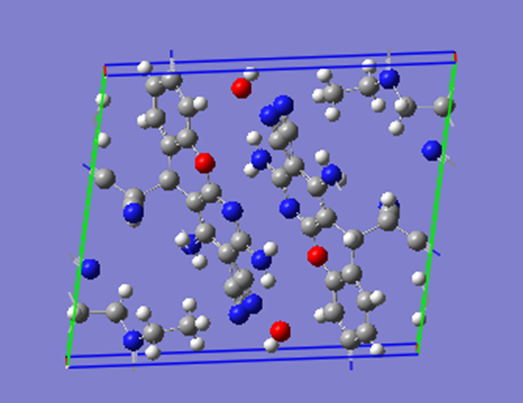
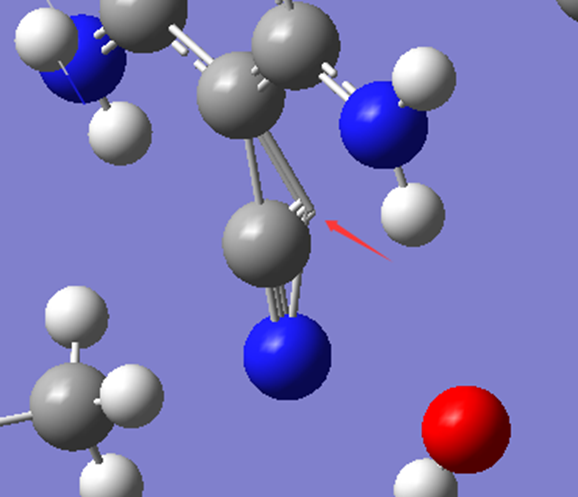
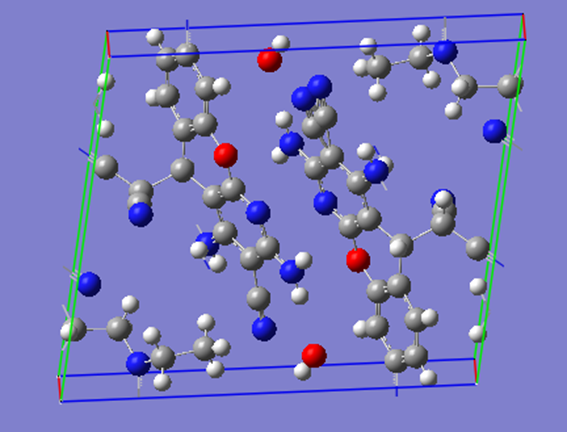
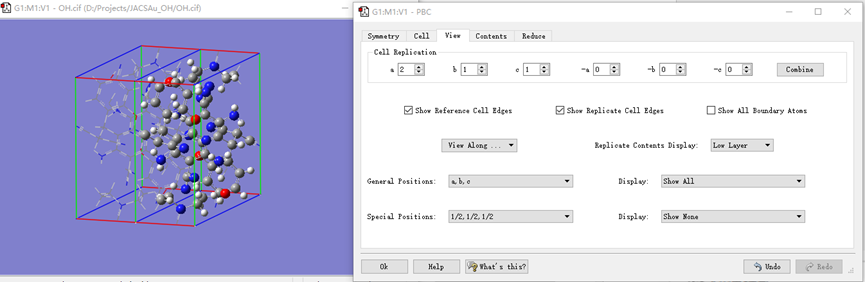
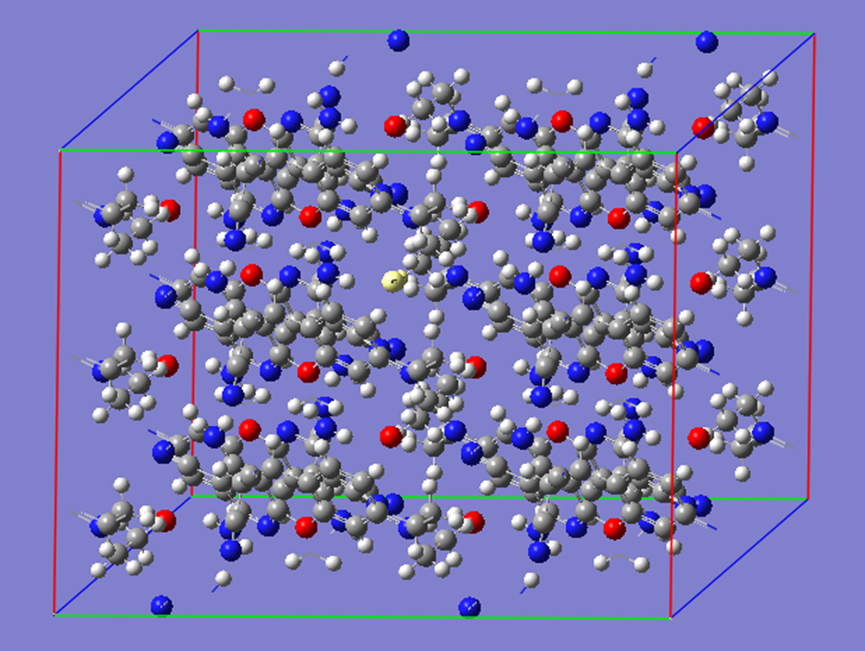
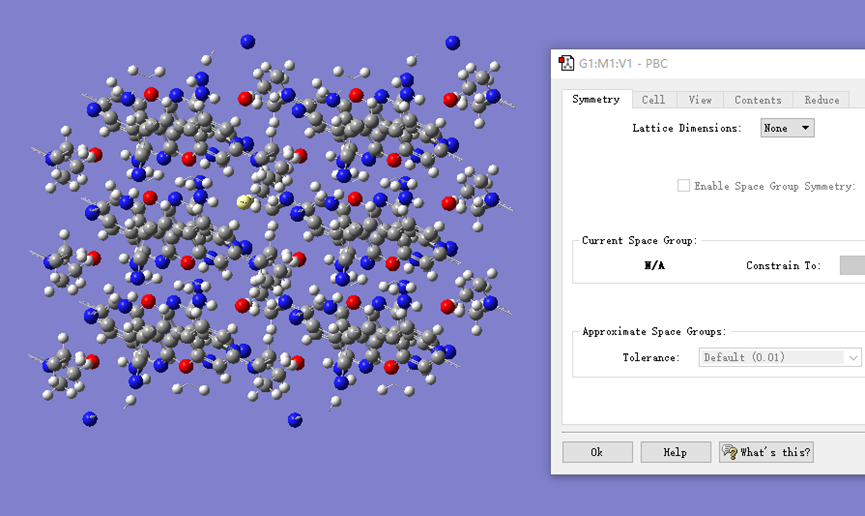
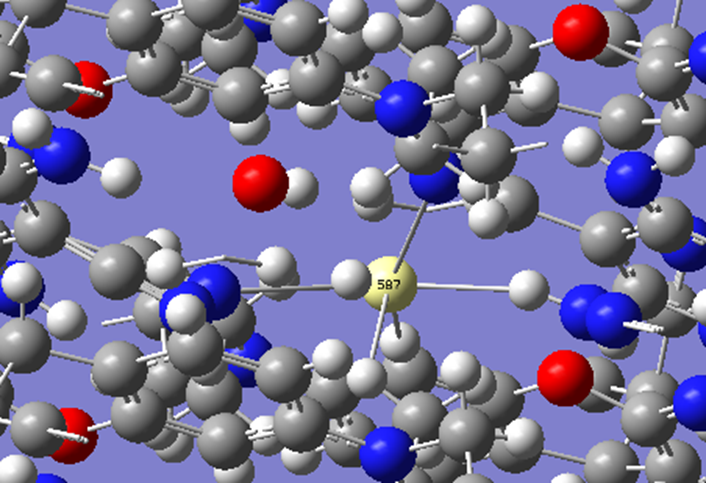
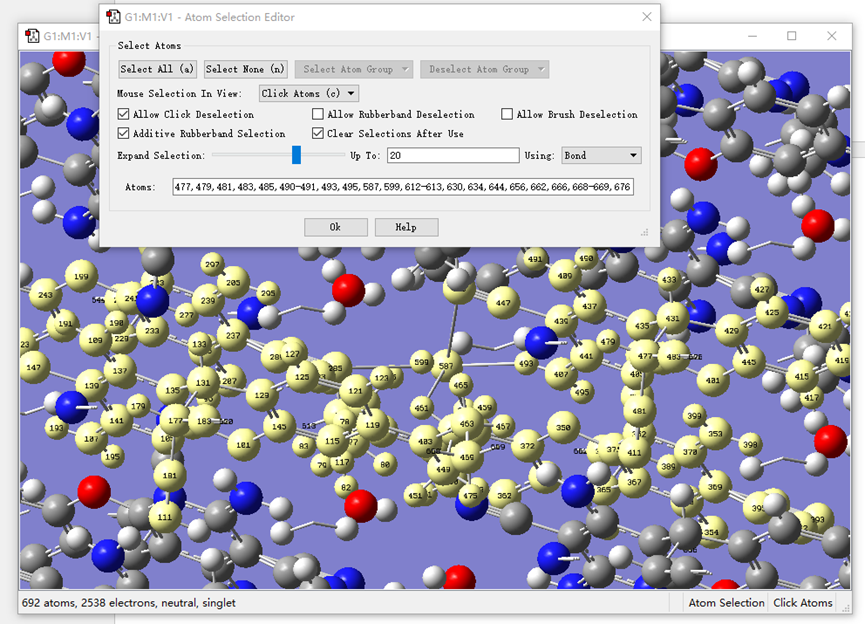
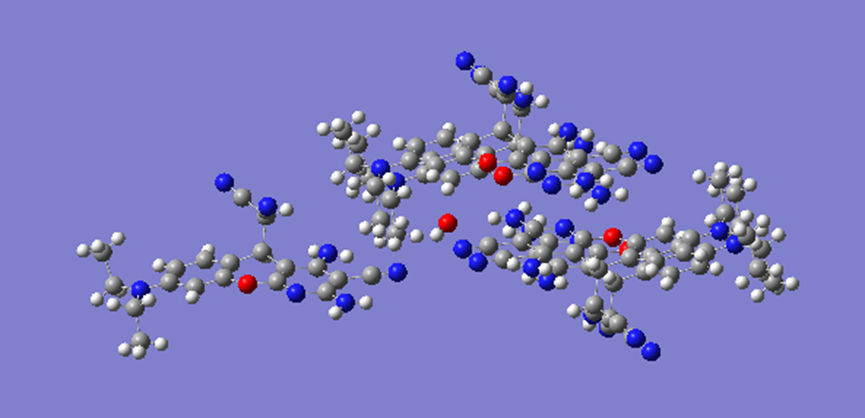
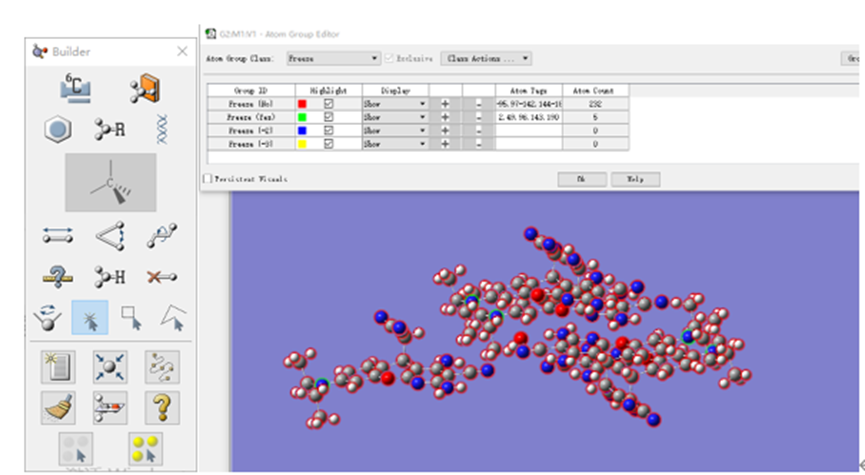
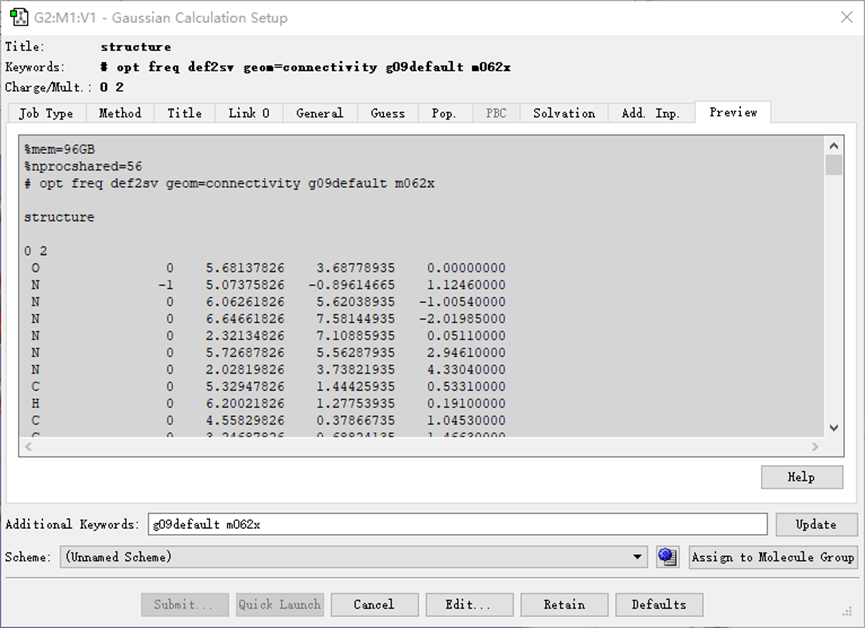
ChevalRita 在DOI:10.1021/jacsau.4c01115中,作者宣称获得了一种包含“OH自由基“的晶体。我们对此很感兴趣,拿到晶体结构细细分析一下。这方面的结果会在另外的帖子里介绍;但我认为这是个很好的例子,可以用来展示如何从文献里扒出晶体结构并建立适合计算分析的模型。 晶体结构的获取 文献中的有机晶体会被收录进CCDC,并获得唯一编号。在JACS Au网站底部可以找到CCDC编号为2351833和2351834,对应文中包含“OH自由基“和乙醇的两种晶体。点击CCDC链接,输入编号: 点击Download selected并同意协议,就可以将两个cif文件下载下来。在CCDC网站上还可以点击View selected进行在线3D结构预览。 我们下载得到的cif文件中记录了选中的两个晶体的信息。用文本编辑器打开后,会发现每个部分遵循相同的格式,开头都有相同的提示框。从#####...处将两部分分开,分别保存成两个cif文件。 这样,我们就获得了两个晶体结构的cif文件,可以用VESTA打开观察。以下是包含EtOH的结构: 可见它相对比较规整,也没有分数占据。 而以下是包含“OH自由基“的结构,比较凌乱。其中,”OH”的占据数是3/4;芳环上氰基的位置没定准,以多个邻近的分数占据原子的方式呈现。 结构的处理 接下来,我们要用这个结构建立适合计算的团簇模型。首先要对结构进行适当的处理,恰当处理分数占据、修整位置不确定的原子。这可以通过结合使用Material Studio和GaussView来完成,也可以只用GaussView进行。两者都是商业软件。在这里,我们介绍单独使用GaussView的方法。 用GaussView打开包含“OH自由基“的结构。占据数会被自动当成1。 在界面处右键View -> Builder,点击删除原子按钮,删除两个氰基中的任意一个。 在删除原子后,还会留下一些“化学键的末端“,也要点击把它们删掉,否则输入文件中会出现一些无法识别的?s行。 删除干净之后,接下来要进行扩胞。扩胞的含义是沿着给定的坐标轴让晶胞重复给定次数然后合并到同一个超胞中。由于我们要建立团簇模型进行非周期性计算,来观察OH自由基在晶体环境中能否存在,所以要建立一个以OH为中心、周围一圈可能与之发生相互作用的位置处尽可能保留有机分子的团簇,这需要先将晶胞扩展到足够大,选出感兴趣的部分,再删掉多余部分实现。 右键Tools->PBC,打开周期性设置窗口。在Cell选项卡里可以查看晶胞参数;为了扩胞,打开View选项卡,在Cell Replication里进行设置。当我们在a、b、c中任意一个文本框中填写扩胞倍数后,在界面中会以线模型的方式给出预览,方便我们知道如何设置。 最终,我们选择a方向填写3,b方向填写2,点击Combine,实现扩胞操作。这样得到的晶胞中,有一个OH自由基(高亮标出)周围一圈都有完整的有机分子。 在Symmetry里将Lattice Dimension改成None,点击确定,取消周期性。接下来只需要删除多余的原子即可。 这个工作看似劳心费神,其实有简单方法。GaussView里有根据化学键关系进行选择的功能。为了利用这一便利,我们先把感兴趣的OH与周围所有希望保留的有机分子中任意一个原子用化学键连接起来: 右键Tools -> Atom Selection,在Expand Selection里扩张到极致,可以发现感兴趣的有机分子就被选中了。注意滑块的末端并不是扩张的极限;我们可以在Up to后面输入一个足够大的数字,实现感兴趣区域的完全选中。 接下来点击OK,感兴趣的部分就被完全选中了。按Ctrl+X将选中部分剪切下来,按Ctrl+N新建一个分子,按Ctrl+V粘贴,我们的团簇模型就抽好了: 设置原子固定 这个有机晶体的结构依赖pi-堆积作用维持。抽出来的团簇如果直接进行构型优化,边缘分子肯定会乱跑,因此需要施加适当的固定。固定要尽可能少,在这里我们固定每个有机分子NEt2基团的氮原子。右键Tools -> Atom Groups,把Atom Group Class选到Freeze。用Builder里的选择工具选中要固定的原子(此时为黄色高亮),在Freeze (Yes)后面的加号上单击,它们就被划入了被固定几何坐标的原子之中(此时变成绿色高亮)。 让我们来看一眼输入文件,可以看到在Gaussian输入文件中这种固定是通过在元素符号后面新增一列实现的,被固定的原子被标记为-1,其余原子被标记为0。利用这个团簇模型就可以开展计算研究了。当然,由于这个模型仍然很大(因为我们目前对这个体系中相互作用的性质尚不清楚,需要尽可能多保留可能跟OH发生作用的有机分子。根据计算结果,可能会进行进一步删减),Gaussian算起来费劲,可以把坐标复制出来用ORCA进行计算,这就超出本文范围了。